Forschung
Abteilung Langer
Mitochondrien sind dynamische Stoffwechselorganellen, die an der zellulären Signalübertragung beteiligt sind und sich an unterschiedliche physiologische Anforderungen anpassen. Die Aktivität der Mitochondrien nimmt mit dem Alter ab und eine Funktionsstörung dieser Organellen wird mit zahlreichen altersbedingten Krankheiten in Verbindung gebracht, was die Frage aufwirft: Wie beeinflussen die Mitochondrien die Zellfitness und das Altern? Um die Rolle der Mitochondrien bei der Alterung und altersbedingten Krankheiten zu verstehen, versucht die Abteilung, die molekularen Mechanismen zu definieren, die die Funktion der Mitochondrien im Alter erhalten und die dynamische Anpassung des mitochondrialen Proteoms auf zell- und gewebespezifische Weise steuern. Die Projekte konzentrieren sich auf die Funktion mitochondrialer Proteasen, die sich als zentrale Regulatoren erweisen und die komplexen Interaktionen der Mitochondrien mit ihrer zellulären Umgebung steuern.
Ausgewählte Projekte
Mitochondriale Membrandynamik und Metabolismus

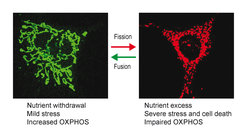
Die Vererbung von Mitochondrien und die Aufrechterhaltung ihrer funktionellen Integrität erfordern, dass Mitochondrien ständig fusionieren und sich teilen. Durch eine ausbalancierte Membranfusion und -spaltung bleibt ein netzartiges, röhrenförmiges Mitochondriennetzwerk erhalten, das sich dynamisch an Stoffwechselbedürfnisse und Stressbedingungen anpasst. Die Fragmentierung dieses Netzwerks ermöglicht die Beseitigung beschädigter Mitochondrien durch Mitophagie und ist mit dem Zelltod verbunden. Dies ist ein Kennzeichen gealterter Zellen und wird bei Krankheiten beobachtet. Die proteolytische Verarbeitung der dynaminähnlichen GTPase OPA1 durch zwei Peptidasen in der inneren Membran der Mitochondrien, OMA1 und YME1L, sorgt für ein Gleichgewicht zwischen Fusion und Spaltung (Anand et al., 2014). Die Spaltung von OPA1 hemmt die mitochondriale Fusion und führt zu einer Fragmentierung des mitochondrialen Netzwerks durch laufende Spaltungsvorgänge. Wir haben beobachtet, dass beide Peptidasen unter verschiedenen physiologischen Bedingungen aktiviert werden. Die proteolytische Verarbeitung von OPA1 durch zwei Peptidasen bildet somit eine Sensoreinheit, die auf unterschiedliche Eingangssignale reagiert und die Anpassung der mitochondrialen Morphologie und Funktion an verschiedene physiologische Anforderungen ermöglicht (MacVicar und Langer, 2016).
OMA1 ist eine stressaktivierte Peptidase (Baker et al., 2014). Die Aktivierung von OMA1 und die mitochondriale Fragmentierung in Kardiomyozyten, denen YME1L fehlt, führt bei Mäusen mittleren Alters zu Zelltod, dilatativer Kardiomyopathie und Herzversagen (Wai et al., 2015). Die mitochondriale Fragmentierung verändert die Brennstoffverwertung der Kardiomyozyten von Lipid zu Glukose, was durch die Ablation von Oma1 unterdrückt werden kann. Dieses verdeutlicht den engen Zusammenhang zwischen mitochondrialer Morphologie und Stoffwechsel. Der Alterungsprozess ist mit weitreichenden Veränderungen des Stoffwechsels verbunden und Stoffwechselinterventionen sind seit langem als wirksame lebensverlängernde Maßnahmen bekannt. Wir wollen daher herausfinden, wie sich Stoffwechselveränderungen auf die mitochondriale Dynamik auswirken und umgekehrt, und die Bedeutung der OMA1-abhängigen mitochondrialen Fragmentierung für das Altern und für altersbedingte mitochondriale Erkrankungen untersuchen.
Mitochondrialer Lipid-Transport und Membran-Homöostase
Die Abnahme der bioenergetischen Kapazität der Mitochondrien mit dem Alter geht mit Veränderungen in der Lipidzusammensetzung ihrer Membranen einher. Dazu gehört die Oxidation von ungesättigten Acylketten der Membranlipide, wie z. B. des mitochondrienspezifischen Phospholipids Cardiolipin. Dieses Peptid ist für die Struktur und Funktion der Mitochondrien sowie für altersbedingte Signal- und Qualitätskontrollwege entscheidend. Wir wissen jedoch nur wenig darüber, wie die Lipidzusammensetzung der Mitochondrienmembranen zeitlich und räumlich reguliert wird. Membranassoziierte Proteasen werden nicht nur von der Membranlipidumgebung beeinflusst, sondern sie regulieren auch selbst das Membranlipidom. Mitochondrien sind Organellen mit Doppelmembran, die viele ihrer Membranlipide aus dem endoplasmatischen Retikulum (ER) erhalten. Phosphatidylethanolamin und Cardiolipin werden jedoch in den Mitochondrien aus Vorläuferlipiden aus dem ER synthetisiert. Wir haben eine neue Klasse von Lipidtransferproteinen im mitochondrialen Intermembranraum identifiziert (Connerth et al., 2012; Potting et al., 2013; Aaltonen et al., 2016; Saita et al., 2018). Diese transportieren Phospholipide zwischen den mitochondrialen Membranen stellen die Phospholipidsynthese in der inneren Membran sicher. Der mitochondriale Lipidverkehr wird streng reguliert, und Veränderungen in der Lipidzusammensetzung der mitochondrialen Membranen haben schwerwiegende Auswirkungen auf die mitochondriale Struktur und Funktion. Wir interessieren uns für die Mechanismen des Lipidtransports in Mitochondrien und arbeiten an einer quantitativen Beschreibung der mitochondrialen Membranhomöostase und ihrer Veränderungen mit dem Alter, indem wir quantitative lipidomische und proteomische Ansätze kombinieren.

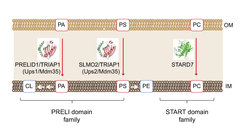
Ein weiterer Schwerpunkt unserer Forschung sind die Membrangerüste der Prohibitin-Familie, die funktionelle Membrandomänen in der inneren Membran organisieren, die durch eine definierte Protein- und Lipidzusammensetzung gekennzeichnet sind (Tatsuta and Langer, 2017). Der gewebespezifische Verlust von Prohibitinen löst altersbedingte Krankheiten wie Neurodegeneration, Kardiomyopathie oder Diabetes aus, was die Bedeutung einer definierten räumlichen Membranorganisation für die funktionelle Integrität der Mitochondrien unterstreicht.
Mitochondriale Dysfunktion und Neurodegeneration
")
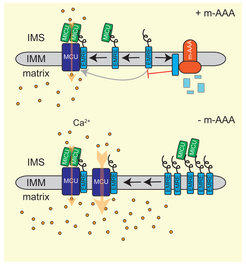
Das Altern ist mit einem erhöhten Risiko für neurodegenerative Erkrankungen verbunden. Das Überleben der Neuronen hängt entscheidend von den Mitochondrien ab und ihre Funktionsbeeinträchtigung bei Krankheiten betrifft vor allem das Gehirn und das Muskelgewebe (Quiros et al., 2015). Ein Beispiel dafür ist die mitochondriale m-AAA-Protease, ein ATP-abhängiger proteolytischer Komplex in der inneren Membran. Mutationen in den Untereinheiten dieser Protease, AFG3L2 und SPG7/Paraplegin, führen zu spinozerebellarer Ataxie (SCA28) bzw. hereditärer spastischer Paraplegie (HSP7). m-AAA-Proteasen üben in den Mitochondrien Qualitätskontroll- und Regulationsfunktionen aus. Die Analyse des neuronalen Proteoms in AFG3L2-defizienten Mäusen ergab, dass m-AAA-Proteasen den Aufbau des mitochondrialen Kalzium-Uniporters MCU regulieren und damit die mitochondriale Kalzium-Homöostase steuern (König et al., 2016). Wir interessieren uns für die pathogenen Mechanismen neurodegenerativer Erkrankungen, die durch eine Störung der mitochondrialen Proteostase verursacht werden, und wollen die auffällige Zell- und Gewebespezifität dieser altersassoziierten Krankheiten entschlüsseln.