Research
Department Langer
Mitochondria are dynamic metabolic organelles that participate in cellular signaling and adapt to varying physiological demands. The activity of mitochondria declines with age and a dysfunction of these organelles is associated with numerous age-associated diseases, raising the question: how do mitochondria influence cell fitness and ageing? In order to understand the role of mitochondria in ageing and age-associated disease, the department seeks to define molecular mechanisms that preserve mitochondrial function with age and drive the dynamic adaptation of the mitochondrial proteome in a cell- and tissue-specific manner. Projects focus on the function of mitochondrial proteases that are emerging as central regulators and orchestrate complex interactions of mitochondria with their cellular environment.
Selected projects
Mitochondrial membrane dynamics and metabolism

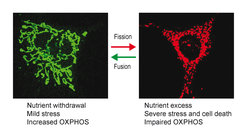
The inheritance of mitochondria and the maintenance of their functional integrity require mitochondria to constantly fuse and divide. Balanced membrane fusion and fission events preserve a reticulated, tubular network of mitochondria that dynamically adapts to metabolic needs and stress conditions. Fragmentation of this network allows removal of damaged mitochondria by mitophagy and is associated with cell death. It is a hallmark of aged cells and is observed in disease. Proteolytic processing of the dynamin-like GTPase OPA1 by two peptidases in the inner membrane of mitochondria, OMA1 and YME1L, balances fusion and fission events (Anand et al., 2014). OPA1 cleavage inhibits mitochondrial fusion, causing fragmentation of the mitochondrial network by ongoing fission events. We observed that both peptidases are activated under different physiological conditions. The proteolytic processing of OPA1 by two peptidases thus forms a sensor unit which responds to different input signals and allows the adaption of mitochondrial morphology and function to various physiological demands (MacVicar and Langer, 2016).
OMA1 is a stress-activated peptidase (Baker et al., 2014). OMA1 activation and mitochondrial fragmentation in cardiomyocytes lacking YME1L induces cell death, dilated cardiomyopathy and heart failure in mid-aged mice (Wai et al., 2015). Mitochondrial fragmentation alters the fuel utilization of cardiomyocytes from lipid to glucose, which can be suppressed by ablation of Oma1, highlighting the intimate link between mitochondrial morphology and metabolism. Ageing is associated with extensive metabolic changes and metabolic interventions are long recognized as potent lifespan extending measures. We are therefore driven to define how metabolic changes affect mitochondrial dynamics and vice versa and to examine the relevance of OMA1-dependent mitochondrial fragmentation for ageing and in age-associated mitochondrial disease.
Mitochondrial lipid trafficking and membrane homeostasis
The decline of the bioenergetics capacity of mitochondria with age is accompanied by alterations in the lipid composition of its membranes. This includes the oxidation of unsaturated membrane lipid acyl chains, such as the mitochondria-specific phospholipid cardiolipin, which is crucial for mitochondrial structure and function as well as age-related signalling and quality control pathways. However, we only have limited understanding of how the lipid composition of mitochondrial membranes is regulated in time and space. Membrane-associated proteases are not only influenced by the membrane lipid environment but they themselves also regulate the membrane lipidome.
Mitochondria are double membrane-bound organelles that receive many of their membrane lipids from the endoplasmic reticulum (ER). Phosphatidylethanolamine and cardiolipin, however, are synthesized within mitochondria from ER-derived precursor lipids. We have identified a novel class of lipid transfer proteins in the mitochondrial intermembrane space (Connerth et al., 2012; Potting et al., 2013; Aaltonen et al., 2016; Saita et al., 2018) that shuttle phospholipids between mitochondrial membranes and ensure phospholipid synthesis in the inner membrane. Mitochondrial lipid trafficking is tightly regulated and alterations in the lipid composition of mitochondrial membranes severely impact on mitochondrial structure and function. We are interested in the mechanisms of lipid transport in mitochondria and work towards a quantitative description of mitochondrial membrane homeostasis and its alterations with age by combining quantitative lipidomic and proteomic approaches.

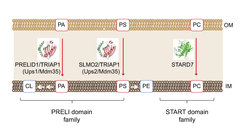
Another focus of our research are membrane scaffolds of the prohibitin family, which organize functional membrane domains in the inner membrane that are characterized by a defined protein and lipid composition (Tatsuta and Langer, 2017). The tissue-specific loss of prohibitins triggers age-related pathologies such as neurodegeneration, cardiomyopathy or diabetes, highlighting the importance of a defined spatial membrane organization for the functional integrity of mitochondria.
Mitochondrial dysfunction and neurodegeneration
")
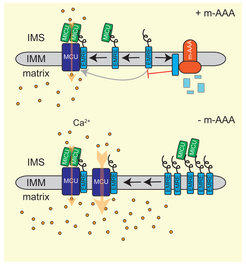
Ageing is associated with an increased risk for neurodegenerative disorders. Neuronal survival critically depends on mitochondria and their functional impairment in disease predominantly affects the brain and muscular tissues (Quiros et al., 2015). Similarly, loss of mitochondrial proteases often results in neurodegeneration, as exemplified by the mitochondrial m-AAA protease, an ATP-dependent proteolytic complex in the inner membrane. Mutations in subunits of this protease, AFG3L2 and SPG7/paraplegin, cause spinocerebellar ataxia (SCA28) and hereditary spastic paraplegia (HSP7), respectively. m-AAA proteases exert quality control and regulatory functions in mitochondria. The analysis of the neuronal proteome in AFG3L2-deficient mice revealed that m-AAA proteases regulate the assembly of the mitochondrial calcium uniporter MCU and thus control mitochondrial calcium homoestasis (König et al., 2016). We are interested in the pathogenic mechanisms of neurodegenerative disorders caused by disturbance in mitochondrial proteostasis and want to unravel the striking cell- and tissue-specificity observed in these age-associated diseases.